商机详情 -
浙江疫苗产品支原体检测验证菌株
来源:
发布时间:2025年11月13日
支原体检测中,污染引发的假阳性是生物制品企业的痛点。实验室环境控制不当、人员操作不规范、仪器耗材混用等因素,都可能导致核酸气溶胶污染或上样污染,进而影响检测结果。一旦出现假阳性,企业需花费大量时间排查验证,严重时会导致批次报废、影响其他产品正常生产。常规 NAT 检测法虽比传统培养法快速,但仍存在明显短板:需配备前处理设备、PCR 仪器等多重耗材,核酸提取、PCR 上机、高浓度产物处理等多个环节均有污染风险;实验准备需 40-60 分钟,操作耗时超 4 小时,每天只能完成 1-2 轮单一类型样本检测,且对场地和人员技能要求极高,需单独划分试剂准备区、样品制备区等多个区域,人力与场地成本高昂。
USP<77> 草案新增支原体 NAT 法方法学验证章节,明确检测限需≥24 次数据支撑统计分析。浙江疫苗产品支原体检测验证菌株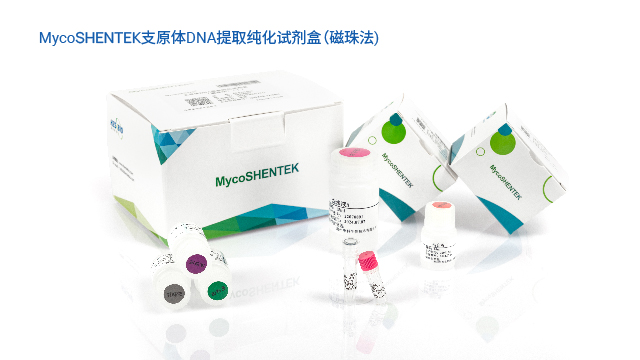
支原体 NAT 检测中异常曲线的出现多与操作设置、耗材使用或实验环境相关,需针对性排查解决。首先需确认软件设置正确性,对照试剂盒说明书检查时间、温度、循环数、荧光采集等参数,尤其需注意关闭不含 ROX 试剂的参比荧光功能。其次若扩增曲线无抬升却出现 CT 值,需调整基线范围,将起点设为荧光信号稳定的循环数,终点设为扩增曲线起峰前一个循环数。再者需确保耗材与仪器匹配,避免使用普通 PCR 耗材或与加热模块规格不符的反应管,防止荧光传导不佳或热传导不充分。此外,曲线先上升后下降可能是反应液蒸发导致,上机前需检查八连管管盖无缝隙,避免液面下降干扰结果。
浙江疫苗产品支原体检测验证菌株支原体检测 NAT 法验证报告需包含检测限、专属性、耐用性等完整数据。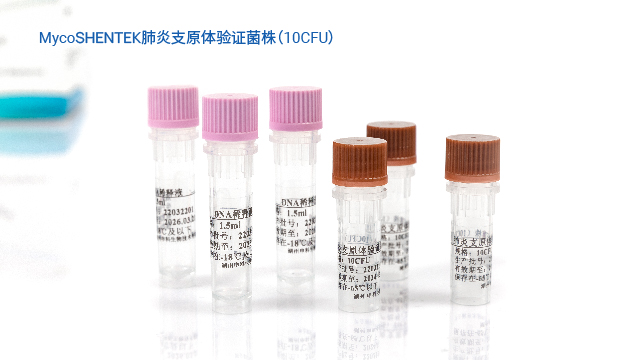
质控结果是支原体 NAT 检测可靠性的前提,需严格遵循判定标准,且需结合实验室实际条件验证适配的标准阈值。质控样品的判定规则明确:NTC(无模板对照)的 FAM 信号需 2 复孔 Ct≥40 或无明显扩增曲线,VIC 信号需 2 复孔 Ct<35 且呈有效 “S” 型扩增;NCS(阴性对照)判定标准与 NTC 一致;PC(阳性质控)的 FAM 信号需 2 复孔 Ct<35 且呈有效 “S” 型扩增,PCS(阳性对照底物)判定标准与 PC 一致。只有质控结果全部满足要求,才能进一步分析样本结果,若质控不达标,需排查设备、试剂、操作等环节的问题并重新检测。
取样量的科学设计直接影响支原体检测的代表性与灵敏度,需结合样品总体积、基质特性综合考量。法规建议:产品总体积大于1000mL的按无菌检查法取样,10-1000mL 取总体积的 1%,1-10mL 取 100μL,小于1mL的需采用替代取样方案。实际检测中,取样体积越大,灵敏度越高,但需平衡洗脱液消耗与重复检测需求。此外,支原体兼具胞内胞外生存特性,只检测上清会导致漏检,需采用细胞裂解等处理方式,确保覆盖全部污染靶点,提升检测结果的真实性。
湖州申科支原体检测验证菌株涵盖 10 余种,满足不同检测场景验证需求。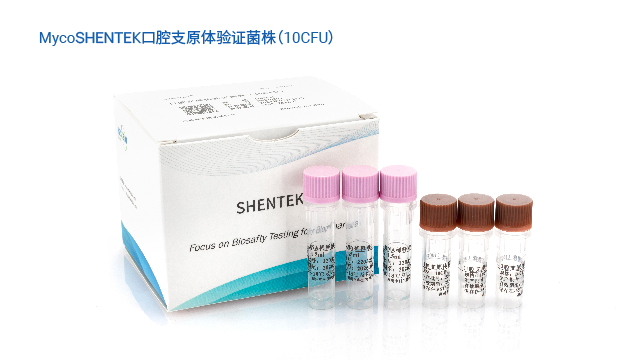
AdvSHENTEK外源因子全自动核酸检测分析系统凭借优越性能,为支原体检测提供坚实技术支撑。硬件方面,系统温度运行精度≤0.5℃,温度波动控制在 ±0.5℃以内,荧光强度 CV≤3%,确保检测结果的重复性与准确性;4 通道单独运行且支持同步检测,通道可叠加延展,兼顾检测效率与灵活性。软件方面,系统具备三级权限管理、日志审计追踪功能,完全符合 21CFR Part11 法规要求,支持 LIMS 系统连接、USB 数据导出及打印机直接打印,满足企业合规追溯需求。此外,系统运输与储存便捷,仪器可常规运输,检测试剂盒在 2-8℃环境下即可稳定保存,无需特殊冷链条件,进一步提升了产品的实用性与适配性。
支原体检测需验证专属性,避免与革兰氏阳性菌交叉反应,确保结果准确。浙江疫苗产品支原体检测验证菌株支原体检测过程中,每批次培养基需做灵敏度测试,确保检测有效。浙江疫苗产品支原体检测验证菌株
为解决传统方法的局限,各国药典纷纷认可支原体核酸检测(NAT)法作为替代方案。EP 2.6.7、USP <77>、JP <G3-14-170>均明确了 NAT 法的应用标准,《中国药典》3301 也规定 “可采用经国家药品检定机构认可的其他方法”,并明确 NAT 法替代培养法时检测限需达 10 CFU/mL,替代指示细胞培养法时需达 100 CFU/mL。NAT 法凭借检测速度快、特异性强的优势,成为新型生物制品支原体检测的理想选择,且法规明确只要通过相关验证,即可正式替代传统方法,为企业提供了合规且高效的检测路径。
浙江疫苗产品支原体检测验证菌株
上一篇:
山东重组药物支原体检测NAT法
下一篇:
四川抗体药物热原检测