商机详情 -
抗体药物宿主细胞残留DNA检测
来源:
发布时间:2025年08月08日
宿主细胞残留DNA检测的样品处理环节是决定检测准确性的关键步骤,其技术难点贯穿于核酸释放、纯化和去除干扰物的全过程。生物制品基质复杂多样,不同类型样品(如疫苗、单抗、干细胞、免疫细胞类产品)对处理方法的要求差异明显:疫苗样品常含高浓度灭活剂(如甲醛)和佐剂(如铝盐),易导致DNA吸附或降解;单抗样品中的高蛋白浓度(10-100 mg/mL)会竞争性结合磁珠,降低提取效率;干细胞、免疫细胞类产品则可能残留二甲基亚砜(DMSO),直接抑制PCR反应。
方法验证是宿主细胞残留 DNA 检测的重要环节,含线性、专属性等指标。抗体药物宿主细胞残留DNA检测
SHENTEK® E1A 残留 DNA 检测试剂盒用于定量检测生物制品中宿主细胞,如HEK293、293T 细胞,来源的 E1A 残留 DNA 的试剂盒。试剂盒利用荧光探针原理,采用 qPCR 方法定量检测样品中 E1A 残留 DNA。检测快速,专一性强,性能稳定可靠。试剂盒配套有 E1A 线性化定量参考品与非线性化定量参考品,供客户针对自己的需求进行选择。本试剂盒与 SHENTEK®宿主细胞残留 DNA 样本前处理试剂盒配套使用,可准确定量样品中残留的微量 E1A DNA。整个分析系统通过优化前处理与检测步骤的兼容性,明显提升对E1A细胞微量 DNA 残留的回收率与定量准确度。
吉林MDCK宿主细胞残留DNA检测基因治疗领域,检测 HEK293 细胞等残留 DNA 及质粒 DNA,规避致瘤性等风险。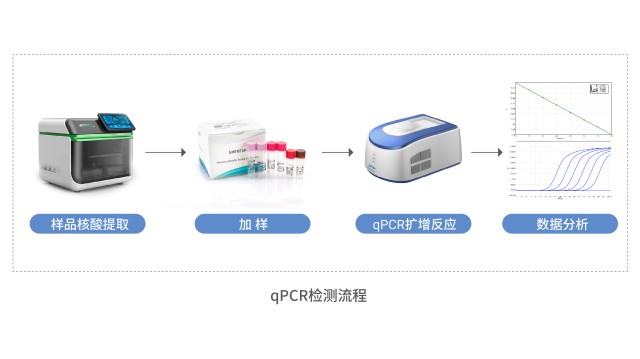
关于宿主细胞残留DNA(rDNA)的风险研究,主要包括传染性、致病性、免疫原性等,各国药典对其残留量有着严格的限度要求:美国食品药品监督管理局(FDA)发布的指导原则中指出生物制品宿主细胞DNA残留限度不得超过100 pg/剂,对于大剂量的生物制品(如单克隆抗体),根据其残留DNA来源及给药途径,DNA残留量可放宽至10 ng/剂。《欧洲药典》通则规定的生物制品残留DNA限度大多为不超过10 ng/剂。2025版《中国药典》三部规定,以细胞基质生产的生物制剂DNA残留量不能超过100 pg/剂。
SHENTEK® Hi5&AcNPV 残留 DNA 检测试剂盒(多重 PCR-荧光探针法)用于定量检测昆虫细胞(High Five)杆状病毒表达系统来源的基因工程疫苗中残留的 Hi5 细胞 DNA和杆状病毒(AcNPV)DNA 的试剂盒。本试剂盒利用荧光探针原理,采用多重 qPCR 的方法定量检测样品中 Hi5 和 AcNPV残留 DNA。检测快速,专一性强,性能稳定可靠,检测限可以达到 50 copies/反应。试剂盒配套有 Hi5&AcNPV 定量参考品。本试剂盒与 SHENTEK®宿主细胞残留 DNA 样本前处理试剂盒配套使用,可准确定量样品中残留的 Hi5&AcNPV 微量 DNA。
宿主细胞残留DNA检测的阴性对照需包含无模板对照(NTC)和阴性样品基质对照,排除假阳性。
宿主细胞残留 DNA 检测存在多种常见问题。首先,扩增异常表现为扩增曲线异样,扩增效率不达标,检测值也会出现异常;第二,回收异常指回收过程有状况,回收率偏高或偏低;第三,污染问题是 NTC(无模板对照 )、NCS(阴性对照 )出现有值情况,干扰检测;第四,设备使用异常则因前处理设备、PCR 设备操作不当,致使检测结果异常。这些问题从扩增、回收、污染到设备操作,多环节影响检测准确性,需在实验中针对性排查、解决,保障宿主细胞残留 DNA 检测结果可靠 。
在CGT领域,对干细胞等产品检测残留 DNA,确保质量可控并符合监管要求。河北生物制品宿主细胞残留DNA检测常见问题SHENTEK® 系列宿主细胞残留DNA检测试剂盒冻融 5 次,性能不受影响。抗体药物宿主细胞残留DNA检测
湖州申科生物疫苗制剂宿主细胞残留DNA样本前处理试剂盒的核酸提取纯化为磁珠法,针对疫苗制剂的产品特点,优化了试剂配方,用于疫苗成品,如Vero细胞生产的狂犬病疫苗中的Vero残留DNA的样品前处理,可稳定高效地获得样品中的痕量DNA,可与各个SHENTEK®宿主细胞DNAqPCR检测试剂盒配合使用。本试剂盒可以通过 rHCDpurify®前处理系统实现样品的自动处理。如样品中含有铝佐剂或右旋糖苷,具体处理方法可咨询湖州申科生物技术股份有限公司。
抗体药物宿主细胞残留DNA检测
上一篇:
江西CHO宿主细胞残留DNA检测生产企业
下一篇:
广东汉逊酵母宿主细胞残留DNA检测