商机详情 -
珠海微量荧光PCR哪家好
聚合酶链式反应PCR技术的基本原理类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性-退火-延伸三个基本反应步骤构成:模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;引物的延伸:DNA模板-引物结合物在72℃、DNA聚合酶(如TaqDNA聚合酶)的作用下,以dNTP为反应原料,靶序列为模板,按碱基互补配对与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链,重复循环变性-退火-延伸三过程就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍。PCR的很大特点是能将微量的DNA大幅增加。珠海微量荧光PCR哪家好

聚合酶链式反应准备:PCR引物设计:PCR反应中有两条引物,即5′端引物和3′引物。设计引物时以一条DNA单链为基准(常以信息链为基准),5′端引物与位于待扩增片段5′端上的一小段DNA序列相同;3′端引物与位于待扩增片段3′端的一小段DNA序列互补。引物设计的基本原则:引物长度:15-30bp,常用为20bp左右。引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C 过多易出现非特异条带。ATGC很好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列参照。温州微量Real-time PCR应用聚合酶链式反应中的DMSO、甘油等,主要用来保持酶的活性和帮助DNA解除缠绕结构。
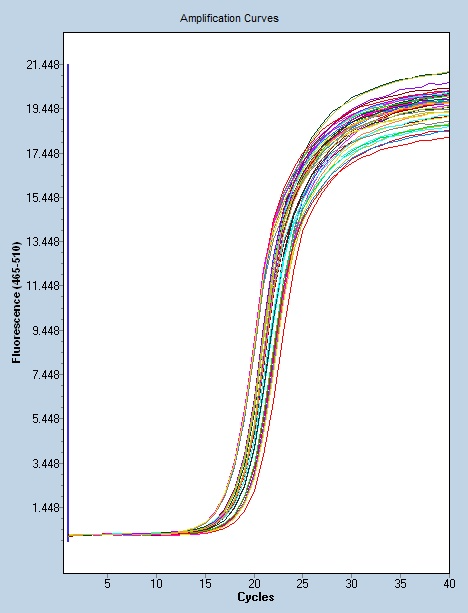
聚合酶链反应:在检测病原体方面,病原体培养是临床诊断的金标准 .但是对于一些苛养菌 生长缓慢的细菌或难以培养的病原体等培养的阳性检出率不高.检测时间过长 无法满足临床早期快速诊断以指导医治的需要[3]。当前 PCR 技术是在传统 PCR 技术应用的基础上进行的改进,包括实时定量 PCR 技术 、 实时荧光定量PCR、 PCR-酶联免疫吸附试验 、 巢式PCR等。 在PCR技术的辅助作用下,实验室检测也逐渐从生化免疫诊断过渡到基因诊断,生化免疫检测主要从表型表现评估病症,而基因诊断则深入本质探究其病因,以PCR 技术为主的核酸技术在临床实验室检测与疾病诊断中表现出了明显的应用价值,在未来的临床医学检验中必将会得到更加较广的应用。
聚合酶链反应的常见问题分析与解决方法:扩增产物出现多条带(杂带):引物用量偏大,引物的特异性不高。应调换引物或降低引物的使用量。循环的次数过多。适当增加模板的量,减少循环次数。酶的用量偏高或酶的质量不好,应降低酶量或调换另一来源的酶。退火温度偏低,退火及延伸时间偏长。应提高退火温度,减少变性与延伸时间,也可采用二种温度的PCR扩增。以2度为梯度设计梯度PCR反应优化退火温度。 样品处理不当。Mg2+浓度偏高,因适当调整Mg2+使用浓度。若为PCR试剂盒,也可能时试剂盒本身质量有问题。复制提前终止。使用非热启动的聚合酶时常有发生。冰上准备反应体系或采用热启动聚合酶。流聚合酶链反应:一种伪等温PCR方法。
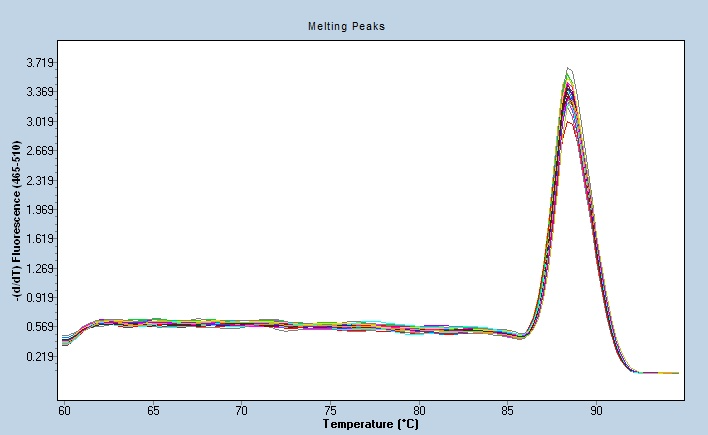
聚合酶链式反应的试验污染:PCR扩增产物污染:这是PCR反应中很主要很常见的污染问题。因为PCR产物拷贝量大(一般为1013拷贝/ml),远远高于PCR检测数个拷贝的极限,所以极微量的PCR产物污染,就可形成假阳性。还有一种容易忽视,很可能造成PCR产物污染的形式是气溶胶污染。在空气与液体面摩擦时就可形成气溶胶,在操作时比较剧烈地摇动反应管,开盖时、吸样时及污染进样的反复吸样都可形成气溶胶而污染。据计算一个气溶胶颗粒可含48000拷贝,因而由其造成的污染是一个值得特别重视的问题。PCR产物的电泳检测时间一般为48h以内,有些很好于当日电泳检测,大于48h后带型不规则甚至消失。珠海微量荧光PCR哪家好
PCR的另一个限制是,即使是很少量的污染DNA也可以被扩增,导致误导或模糊的结果。珠海微量荧光PCR哪家好
现在有些PCR因为扩增区很短,即使Taq酶活性不是很好也能在很短的时间内复制完成,因此可以改为两步法,即退火和延伸同时在60℃-65℃间进行,以减少一次升降温过程,提高了反应速度。检测:PCR反应扩增出了高的拷贝数,下一步检测就成了关键。荧光素(溴化乙锭,EB)染色凝胶电泳是很常用的检测手段。电泳法检测特异性是不太高的,因此引物两聚体等非特异性的杂交体很容易引起误判。但因为其简捷易行,成为了主流检测方法。近年来以荧光探针为的检测方法,有逐渐取代电泳法的趋势。珠海微量荧光PCR哪家好